Kiwa is een autoriteit in veel verschillende markten, van de bouw- en energiesector tot de watersector en de gezondheidszorg – en dat over de hele wereld. Onze experts weten alles over de Europese en wereldwijde regelgeving, kwaliteitsnormen en veiligheidscriteria. Met deze kennis gaan er deuren voor u open die anders gesloten blijven. Wij kunnen u helpen met de toegang tot bepaalde markten. En we kunnen veel voor u certificeren.
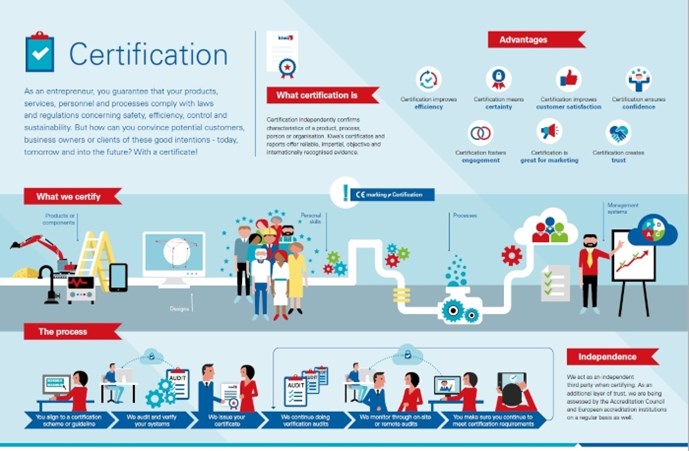
Certificering van een product of een aspect ervan
Met productcertificering kunt u laten zien dat u intern de kwaliteit controleert en beheert in elk onderdeel van het productieproces, van het controleren van de aangeleverde grondstoffen tot het opslaan van het eindproduct. Voor de certificering van uw product controleren wij in feite uw interne systeem voor kwaliteitscontrole. Zo voldoet uw product altijd aan de eisen. Met productcertificering vergroot u uw kansen op de markt doordat u (potentiële) klanten een objectief bewijs kunt leveren dat uw producten en het productieproces aan de geldende kwaliteits- en veiligheidseisen voldoen.
Misschien wilt u niet een ‘heel’ product certificeren, maar alleen een bepaald aspect of kenmerk ervan. Dan kunnen wij een aspectcertificaat leveren. Bijvoorbeeld om te verklaren dat uw product veilig kan worden aangesloten op een drinkwaterinstallatie.
Certificering van een bouwontwerp
Een bouwattest is een eenmalige beoordeling van een bouwontwerp. Een attest doet geen uitspraken over producteigenschappen, hoewel het gekoppeld kan worden aan de certificering van een systeem of product. Een attest bewijst alleen dat een bouwelement aan de prestatie-eisen voldoet zoals deze zijn opgesteld in het bouwbesluit van een land of een gelijkwaardig document.
Certificering van persoonlijke vaardigheden
Mensen zijn de belangrijkste factor bij het bereiken van uw bedrijfsdoelen. Certificering van personen, of liever gezegd, van persoonlijke vaardigheden, vormt een aanvulling op de kwaliteitsborging van organisaties en producten. In tegenstelling tot onderwijsprogramma's, die vooral gericht zijn op het verwerven van bepaalde kennis, richten wij ons op het vaststellen van iemands vaardigheden en vervolgens op het controleren ervan. Nadat de geldigheidsperiode van een persoonscertificaat is verlopen, moet de betreffende persoon aantonen dat hij of zij nog steeds over de voor zijn of haar expertisegebied vereiste vaardigheden beschikt.
Certificering van een proces
Als u een service levert of iets produceert, beheert of verwerkt dat niet tegen objectief meetbare criteria getest kan worden, kan Kiwa het werkproces certificeren. Dit geldt ook voor processen op verschillende locaties en processen waarbij vakmanschap een rol speelt. De doorlopende controle van de kwaliteit van alle mogelijke activiteiten in uw organisatie verbetert de efficiëntie en de nauwkeurigheid van medewerkers en maakt het beheersen van bedrijfsrisico's gemakkelijker. Voorbeelden van dergelijke processen zijn het aanleggen van leidingsystemen, het vervaardigen van funderingspalen en het aanbrengen van vloeistofdichte bestrating bij tankstations. Een procescertificaat versterkt uw positie op de markt en biedt u voordeel bij de toewijzing van projecten.
Certificering van (management)systemen
Systeemcertificering richt zich op het managementsysteem dat u in uw bedrijf of instelling hanteert. Dit brengt de verantwoordelijkheden, autorisaties en procedures in uw organisatie in kaart. De meeste tekortkomingen in een organisatie zijn terug te voeren op onduidelijke communicatie en onduidelijkheid over verantwoordelijkheden, taken, competenties en procedures. Een kwaliteitsmanagementsysteem volgens een internationale ISO-norm maakt mensen bewuster van wat ze doen en hoe, wanneer en waarom ze dat doen. De mening van klanten komt naar voren uit klanttevredenheidsonderzoeken. Met een goed functionerend managementsysteem zijn ook andere aspecten van de bedrijfsactiviteiten beter beheersbaar. De voordelen zijn duidelijk: efficiëntere werkprocessen, blijere medewerkers, betere risicobeheersing, meer klanttevredenheid en een hogere winst.
CE-markering
CE-markering is geen certificaat. Toch is het heel belangrijk als u producten op de Europese markt wilt brengen. CE staat voor Conformité Européenne (Europese Conformiteit). Als fabrikant is het uw verantwoordelijkheid om een conformiteitsbeoordeling uit te voeren, een technisch dossier voor te bereiden, de conformiteitsverklaring op te stellen en de CE-markering op uw product aan te brengen. Als u CE-markering aanbrengt, verklaart u dat uw product is getest en voldoet aan Europese richtlijnen op het gebied van veiligheid, gezondheid en milieu. Deze richtlijnen kunnen voorschrijven dat u uw product laat testen door een bevoegde keuringsinstantie, ook wel een aangemelde instantie genoemd. De EU heeft Kiwa aangewezen als aangemelde instantie; dat betekent dat Kiwa bevoegd is om CE-markering af te geven. Productgroepen waarvoor CE-markering vereist is, zijn bijvoorbeeld machines, gastoestellen, drukapparatuur, persoonlijke beschermingsmiddelen, medische hulpmiddelen, bouwproducten, elektrische en elektronische apparatuur, pleziervaartuigen en speelgoed.
Bekijk de notificaties van Kiwa op de website van NANDO (New Approach Notified and Designated Organisations)